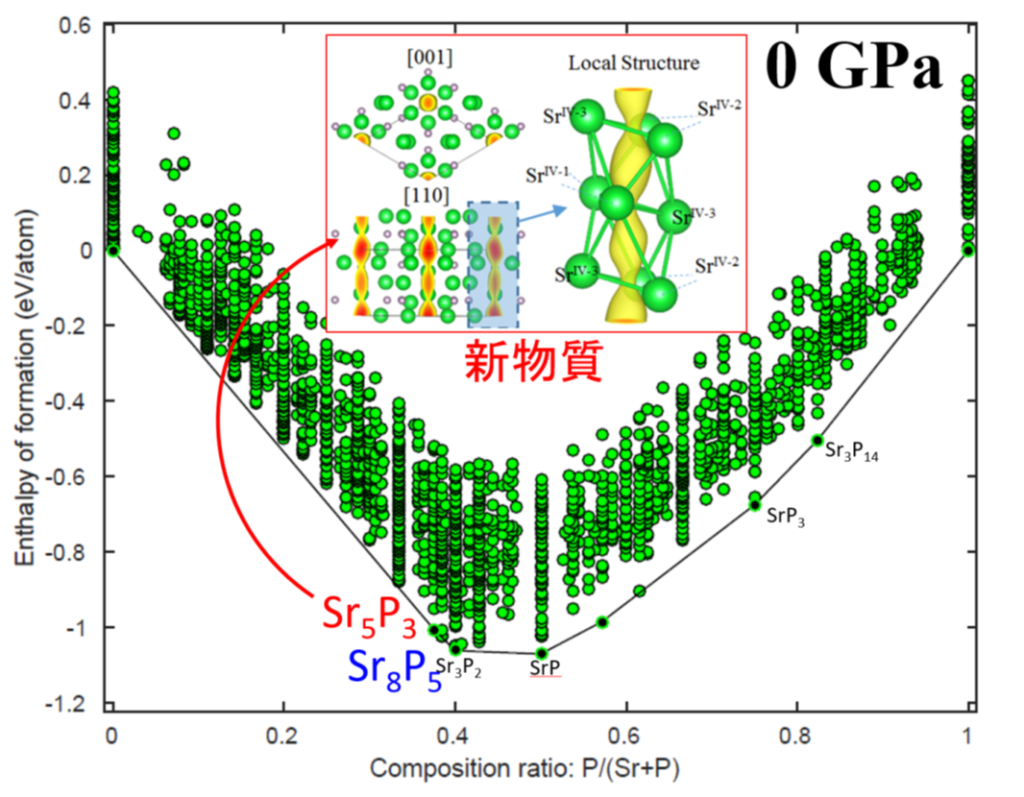
We conduct genetic algorithm structure prediction combined with first-principles electronic structure calculations to determine stable compositions and structures based only on information of elements included in target conpounds. By using this method, it is possible to explore bulk structures, surface structures, sheet structures, nanocluster structures, etc. Examples of our research achievements in this topic are new electrides, new MAX phase compounds containing boron, and two-dimensional compounds.
【Electron transport】
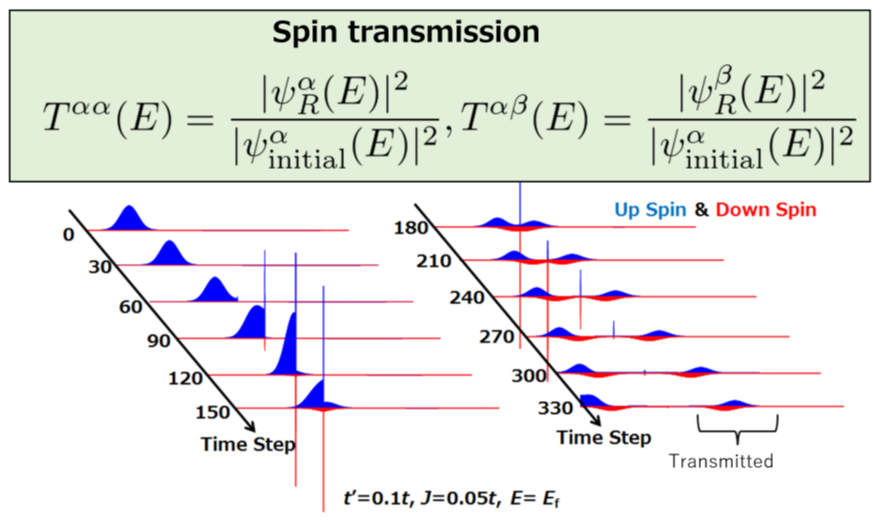
The understanding of electronic states and dynamics in materials is very important not only for the design of electronic devices, but also for the prediction of functional materials. The calculations we conduct in this topic are standard band structure calculations, time propagation of wave functions using the time-dependent Schrödinger equation, and spin-dependent electron tunneling using the non-equilibrium Green’s function method.
【Ionic conduction】
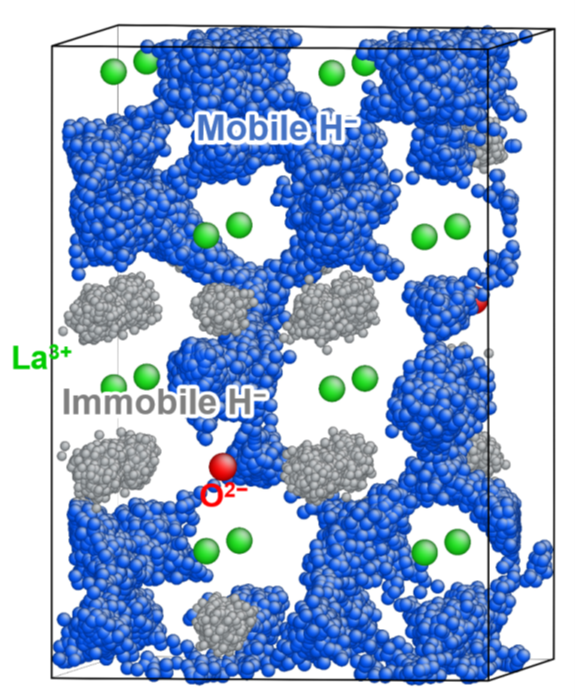
Ionic conduction is a phenomenon in which ions move/diffuse in materials, and is an extremely important process in the development of energy conversion materials. In our laboratory, we are conducting first-principles molecular dynamics calculations, neural-network potential molecular dynamics calculations, and kinetic Monte Carlo calculations. Examples of our research targets in this topic are oxygen ion, proton, and hydride conductors.
【Catalytic reaction】
Chemical reactions are the cornerstone of substance conversion, and catalysts are essential to achieve them efficiently. In our laboratory, we investigate reaction paths by first-principles reaction calculations (transition state) and first-principles molecular dynamics calculations (dynamics). Examples of our research achievements in this topic are polymerization reactions, ammonia synthesis, and decomposition reactions using electrides.
【Device modeling】
It has become possible to calculate the stability, physical properties, and reactivity with sufficient accuracy of materials, but in device simulations, we have to extend the simulation scale in both size and time domains. First-principles calculations are generally limited to hundreds ~ thousands of atoms and picosecond dynamics. We thus are developing a kinetic Monte Carlo method with open boundary conditions for non-equilibrium situation, which enables a large-scale atomistic calculations. By using this method, we can design electrochemical devices in an electrochemical environment by monitoring the device response (DC/AC) dependent on device structures.
【Quantum algorithm】
Although quantum computer is expected to be quite a powerful computer in future, but an algorithm that is constructed for quantum computers (i.e., quantum algorithm) is necessary to realize the very powerful calculations by quantum computer. In our laboratory, quantum algorithms for solid-state material calculations and molecular identification are developed.